
Short Introduction
Protein-folding 예측의 선두주자인 RoseTTA fold와 AlphaFold2의 작동원리를 살펴보고, 관련 알고리즘을 이해하는 활동을 진행하였다.
Content
- 프로젝트 진행 동기
: 예전부터 분자생물학에서 중요한 역할을 담당하는 단백질의 3D structure를 예측하는 것이 고질적인 문제로 자리잡았었다. 단백질의 가장 큰 특징은 단순한 아미노산의 배열이 결정된 순간, 외부적인 요인과 아미노산 간의 상호작용으로 가장 안정된 3D 구조가 존재한다는 것이다. 한편, 아미노산 시퀀스가 주어졌을 때, 직접적으로 3D 구조를 예측하는 것에 어려움을 겪고 있었기 때문에, 최근 들어 급부상한 딥러닝 기반 단백질 접힘 알고리즘이 우후죽순 등장하였다. 그러한 인공지능 중에서도 가장 두각을 드러내는 두 인공지능, AlphaFold2와 RoseTTA fold가 소스코드를 공개했다는 소식을 들었다. 나는 이러한 단백질의 특성에 굉장히 흥미를 느꼈기 때문에 어떠한 원리를 이용해 위의 인공지능을 개발하였는지 궁금해졌다.
- 프로젝트의 주요 내용
: 본 프로젝트에서는 단백질의 3차원 구조를 예측하는 원리가 어떻게 되는지, 그리고 AlphaFold2와 RoseTTA fold가 어떠한 방식을 이용해 3차원 구조를 예측하고 성과를 보였는지, 끝으로 실제로 AlphaFold2를 이용해 Hexokinase라는 효소를 구현해보며, 어느 정도의 정확성을 보이는지 살펴보았다.
- AlphaFold
구글 딥마인드에서 개발한 딥러닝 기반 알고리즘으로, 두 인공지능이 채택한 방식이 다르다는 점에서 주목받을 만하다. 먼저 알파폴드는 합성곱신경망(CNN, Convolutional Neural Network)를 이용해 아미노산 서열은 하나의 이미지라고 받아들여 대응되는 3차원 구조를 형성하는 방식을 채택하였다. 따라서 기존에 구조가 알려져 있는 많은 양의 단백질 데이터베이스를 이용하여 비슷한 아미노산 서열의 순서와 배치를 갖는 단백질을 찾아, 그에 해당하는 부분을 3차원으로 구현하는 것이다. 다만, 이러한 방식은 효소의 활성부위(binding site)를 제대로 표현하지 못하고, 국소적인(local) 단백질의 부위만을 형성시킬 수 있다 보니, 전체적인 단백질의 기능을 예측하는 것에 자주 실패하는 모습을 보였다.
2. AlphaFold2
이에 구글 딥마인드 개발진 측에서는 2021년 새로운 형태의 딥러닝 기반 단백질 예측 알고리즘을 개발하였는데, 그것이 알파폴드2(AlphaFold2)다. 알파폴드2는 어텐션 모듈(Attention Module)과 DNN(Deep Neural Network)을 모두 적용하여 실제 구조와 흡사한 단백질 예측 알고리즘을 개발하게 되었다. 아래는 실제 알파폴드2가 적용시킨 알고리즘의 개요다.
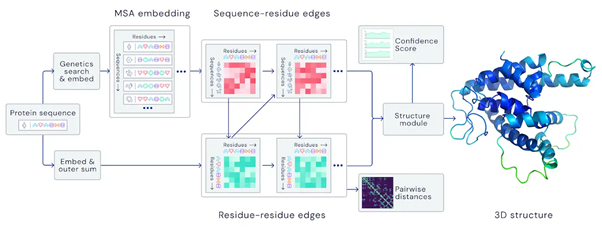
맨처음 인코딩하고 싶은 아미노산 서열이 주어진다면, 이를 이용해 다른 단백질 데이터베이스로부터, 유사한 단백질 시퀀스를 검색한다. 이러한 단백질 시퀀스를 MSA라고 부르며, 얻어진 MSA 데이터셋 간의 데이터 비교와 목표 시퀀스와 MSA 데이터 간의 비교를 attention module를 활용해 유사성을 검토한다. 이 과정을 통해서 structure module을 구할 수 있고, 최종적으로 confidence score를 매김으로 하여금 단백질의 부분별 정확도를 제시하고 있다.
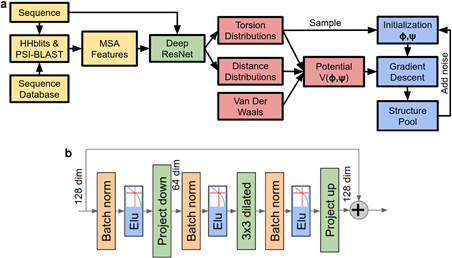
Attention module을 이용해 분석할 때 고려하는 부분에 대하여 조금 더 상세히 설명된 그림이다. 실제로 단백질이 3차원 구조를 이룰 때에는 환경적인 요소와 아미노산 간의 상호작용에 의해서 가장 최적화된 상태가 존재할 수 있는데 이를 위해선 chemical potential Energy값과 더불어 nonpolar interaction 등 다양한 요인을 이용해 계산하여야 한다. 이와 같은 방식으로 얻어진 단백질 역시도 어느 정도 한계가 있는데, 끝부분의 embedding은 제대로 구현하기 힘들고, 금속화 효소와 같은 경우, 금속 이온과의 반응성 역시 적은 편이라는 점을 고려해보아야 한다.
3. Attention module
: 인코더와 디코더를 구분하여, 하나의 context vector를 만든다는 점에서는 Seq2Seq와 비슷하지만, 그것의 단점을 일부 보완한 형태의 인공신경망이다. 기존에 Seq2Seq에서는 context vector를 형성함에 있어서, 하나의 vector에 모든 input data를 정리하여 정보의 손실이 발생하게 된다. 한편, attention module에서는 각 input data에 대하여 encoding을 진행한 뒤, 그들을 순차적으로 decode하는 모습을 보여준다. 즉, attention vector를 형성할 때, 각각의 가중치를 다르게 책정하여 softmax하여 정보의 손실을 최대한 줄이는 방식을 채택했다. 따라서 decoding을 진행함에도 불구하고 Seq2Seq와는 다르게 진행된다.
4. Hexokinase를 활용해 AlphaFold2의 단백질 접힘 구조 확인
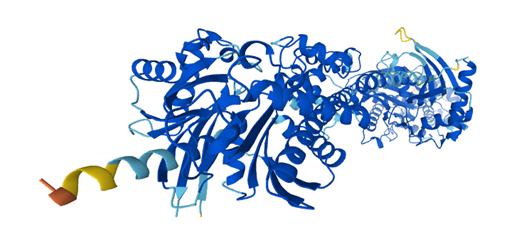
Hexokinase는 대표적인 glycolysis에 관여하는 효소로, glucose로부터 glucose-6-phosphate를 형성시키는 효소이다. 유명한 효소이기에, database와의 연관성을 찾기 훨씬 수월했을 것이라 판단해 hexokinase라는 효소를 이용해 관찰하기로 결정했다. 위 그림에서 파란색에 가까울수록 정확성이 높다는 것을 의미한다. 앞서 소개한 바와 같이 Hexokinase의 경우에도 끝부분에 대한 정확성이 떨어지는 것을 확인할 수 있다.
About
본 글은 2021년도 2학기에 20 강성민 연구회원이 진행한 프로젝트입니다.